
ONT long-read sequencing and Illumina short-read sequencing of 16S rDNA amplicons give comparable results in terms of bacterial community structure in marine sediments
 based on reviews by 2 anonymous reviewers
based on reviews by 2 anonymous reviewers
Fine-scale congruence in bacterial community structure from marine sediments sequenced by short-reads on Illumina and long-reads on Nanopore
Abstract
Recommendation: posted 12 October 2023, validated 13 October 2023
Spor, A. (2023) ONT long-read sequencing and Illumina short-read sequencing of 16S rDNA amplicons give comparable results in terms of bacterial community structure in marine sediments. Peer Community in Microbiology, 100014. https://doi.org/10.24072/pci.microbiol.100014
Recommendation
ONT long-read high-throughput sequencing is not routinely used for metabarcoding studies of microbial communities. Even though this technology is supposed to considerably improve phylogenetic coverage and taxonomic resolution, it initially suffered from relatively poor read accuracy. Assessment of the performance of this new approach in comparison with routinely used 16S rDNA short-read sequencing is therefore needed to validate its use.
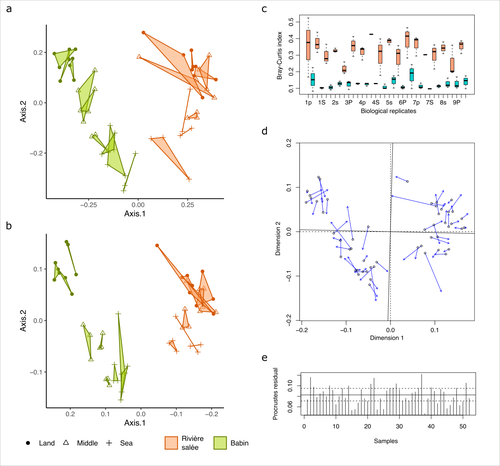
The study by Lemoinne et al. (2023) offers a comprehensive comparison of two 16S rDNA metabarcoding approaches on marine sediment samples. By comparing Illumina short-read sequencing with ONT long-read sequencing, the authors conclude that bacterial community structures inferred from both technologies were similar. They also found that differences observed between sampling sites and along the sea-land orientation were comparable between the two technologies. However, the choice of technology still has an impact on the obtained results, notably in terms of bacterial diversity retrieved, taxonomic resolution, and replicability between biological replicates.
Altogether, these results validate the use of ONT long-read sequencing for 16S metabarcoding approaches in marine sediments. Comparisons of such kinds targeting other remote environments are needed, as they might offer new opportunities for field scientists with no access to sequencing platforms to study the structure and composition of microbial communities.
Reference
Lemoinne, A., Dirberg, G., Georges, M., & Robinet, T. (2023). Fine-scale congruence in bacterial community structure from marine sediments sequenced by short-reads on Illumina and long-reads on Nanopore. biorXiv, version 3 peer-reviewed and recommended by Peer Community in Microbiology. https://doi.org/10.1101/2023.06.06.541006

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
Office Français de la Biodiversité (OFB) and BOREA lab (MNHN)
Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2023.06.06.541006
Version of the preprint: 1
Author's Reply, 12 Sep 2023
Decision by Aymé Spor, posted 17 Jul 2023, validated 18 Jul 2023
Dear authors,
Two reviewers have now evaluated your manuscript and given useful comments for improving its quality.
I suggest you take all the reviewers' comments into account for providing a revised version of the manuscript. More specifically, I would ask you to particularly take into account reviewer 2's comments regarding your experimental design (confounding factors between technologies and primers choice) and elaborate upon this in the revised version.
Sincerely yours,
Aymé Spor
Reviewed by anonymous reviewer 1, 30 Jun 2023
Dear Authors,
thank you for opportunity to review your manuscript. It shows detailed comparison of two sequencing approaches with potentially large impact on microbial alpha diversity of biological samples. Higher number of taxa obtained using long read sequencing is shown after rigorous analysis. On the other hand, similarity of major patterns of community composition between short and long read approach is presented.
Although authors avoid giving strong recommendations on which method should be used, I think that presented results show valuable comparison which is useful for readers oriented on methodological papers.
I listed my line by line comments below. I noted two major issues from which one is about PCR cycling conditions (L152, L158, L173, L182) and the second is about input data into random forest analysis (L311). I ask authors to consider these and other comments below.
Based on this, I think that current version of the manuscript needs minor revisions.
L22, L25 - genera instead of genus
L27 - Here the statement can be stronger, I would omit "probably" since these are real reasons for discrepancies
L60 - please omit "works"
L62 - If there is a length limit for PacBio, it is for sure longer. I suggest to avoid specific number (as it might be obsolete soon) and mention "tens of kbp" or similar
L106, L109 - please omit parentheses at the beginning of sentences
L139 - Please specify type of ZymoBIOMICS mock sample as there are more types on manufacturer's website.
L144 - Starting from this section but also in previous parts of the manuscript, the typograhic corrections need to be applied widely. This includes 16S instead of 16s, dot as decimal point, en dashes where appropriate, English quotation marks, multiplication sign instead of x, etc.
L152, L158 - Altogether 60 cycles of amplification after primary and secondary PCR seems like a lot of cycles. The authors want to avoid PCR biases and do triplicate PCRs (L145) which is certainly good. But then DNA goes through so many cycles which increase chance for bias and contamination amplification. Could you please include reference, if there is such approach recommended?
L163 - Please include full names of sequencing kits as they are written at manufacturer's webpage.
L169 - I agree with usage of specific primers for Archaea, but I am missing explanation of such approach in Introduction or in Methods. Could you please include one sentence why archaeal primers were used in the case of Nanopore?
L173, L182 - Here amplicons for Nanopore went through 55 cycles which posses same question if it is necessary to cycle so many times.
L173, L174 - unclear meaning of values in brackets, please clarify
L177 - diluted is maybe better then reduced
L188 - "protocol from Nanopore website" There is no need to specify from which website the protocol was downloaded. Alternatively, you can include link as proper reference.
L195 - please check the number 1624, L257 and L479 mention different level of rarefaction
L198 - PRJNA985243 checked and fastq files are available together with clear labelling of individual samples
L202, L208 - the connection of ASVs and OTUs is a little bit confusing here. I understand that DADA2 was used to merge pair-end reads (L202) and maybe to correct errors. Individual sequences were then clustered at 97% threshold. If it is so, please clarify the paragraph. It was not ASV table which was clustered (L208) but rather individual sequences, right?
L207 - please correct typo in kpb
L208 - please consider to include vsearch version
L215-218 - The sentence needs clarification, its meaning is unclear.
L230 - Please check, maybe Figure 4 was meant.
L232 - Please specify core threshold. Is it >=50% in each sample, >=50% of all samples or something else?
L240-L243 - The sentence is duplicated, please correct.
Figure 2 - Please include description in figure caption that "once" means one flow-cell (L467) and "twice" means sequenced on two flo-cells (L468).
L250 - I wonder if manufacturer took into account copy number of 16S genes of individual genomes present in mock sample. Taxon with 2 copies of 16S rRNA will show higher relative abundance in final sequences than taxon with one copy. This was probably considered during mock preparation but it might be one of the explanations for changed proportions. However, I am aware that there is nicely looking barplot with even distribution of taxa.
L261 - This is side note, but Table 1 partly duplicates information in Figure 3.
L267 - I suggets to include information what is the mean abundance of 11 phyla detected exclusively in Nanopore data. This might provide an idea about size of this Nanopore-detected subcommunity.
L270 - please omit only
L276 - please consider to reorder Figure 4 and Figure 3. In the current version, Fig 3 is referenced after Fig 4.
L282 and L288 - information here is repeated, please correct
L286 - genera
L296 - L305 - For reader's reference, I suggest to include phylum names of individual orders in brackets.
L309 - Does the "species rank" means that OTUs served as input into Mantel test? Please clarify.
L311 - Genera and families are arbitrary groups and as such I am not sure if they can enter random forest. I suggest to test the same effects with OTUs which are exactly defined.
L315 - The archaeal sentence sounds a little bit vague. I suggest to include at least information on how many phyla were detected as Nanopore-only.
L323 - L325 - nicely summarised output which applies also in this manuscript
L342 - Please consider to add that another reason might be due to incomplete databases
L346 - What do you mean by maximum resolution? I feel that this sentence needs reformulating.
L359 - L361 - I understand what was meant here but I feel that this sentence needs reformulating.
L362-L364 - Nice key output of the study.
L365 - Ecology of Nanopore-only taxa can not be inferred based on the fact that the rest of core community was similar between Nanopore and Illumina. E.g. Nitrospinota detected by Nanopore might represent low-density nitrifiers with potentially high impact on N cycling in sediment.
L368-L380 - The last paragraph seems out of context, please consider mentioning portability in Introduction if you prefer to keep it. I think that manuscript has same quality even without portability section.
Reviewed by anonymous reviewer 2, 11 Jul 2023
The present study compares short read sequencing with long read sequencing from ONT on environmental (marine) sediment samples. The authors conclude in this comparison that ONT works as good as Illumina with even covering more diversity. The articles writing is okay, and the findings are concisely presented. I think the study design as it is presented is however not correct, while the conclusions are partially valid (see below). I have one important methodological question and one important question related to the mock community.
Line 87-88: please give the respective references for RCA and UMI already in this sentence.
Lune 105: you may add https://doi.org/10.1093/femsec/fiac120 to the list. And I think there are also others that are more and more using it.
Line 113: The Study design description is not quite accurate: how do the authors disentangle the effects of primers from the effects of sequencing technologies? This can’t be done with this data. (except maybe with an in silico PCR and Illumina simulation on the Nanopore reads). Therefore, what the authors really compare were short amplicons with primer pair A with long amplicons with primer pair B. Since the study of Parada et al. 2015, we know that even a single nucleotide in one primer can have a tremendous effect on diversity estimates from sequencing. Here, the authors compare different primer sets, which renders a comparison of sequencing technologies not quite on the point. Therefore, it is rather a feasibility study on long read sequencing with ONT that shows, that it produces similar results as established primers for short read sequencers. A direct comparison with numbers (alpha diversity estimates) is not advised, because it is like comparing apples with oranges. Therefore, I am afraid that the aim and the writing of the manuscript needs to revised accordingly (and rather extensively).
Method: LSK109 with 10% error rate; clustering at 97% will result in spurious OTUs, even with singletons across all samples removed
Line 221: I would advise to look into the new publications from Patrick Schloss (doi: https://doi.org/10.1101/2023.06.23.546313) considering this argument, in particular for the alpha diversity estimates, since much weight is put on it in the author’s manuscript
Line 246. Relative proportions of the mock community are one thing, but not really that relevant since we are talking about compositional data. More important is the matching of OTU numbers with actual # of taxa in the Mock community and the detection of all taxa. I can imagine that there are vast differences between the amplicons. Please amend these missing results, even if they represent a weak point for Nanopore R9.
Line 254: This comparison with percentage suggest that more species are better. This is however not the case. An accurate estimate of the taxa in a given sample is important. More OTUs may for instance mean more artifacts, less true taxa.
Line 266: Nanopore detected these 11 phyla or did the primer system detect these phyla. I would argue that the primer pair detected it. ONT is just the tool to read out these sequences. I would suggest to revise this terminology by replacing “Nanopore” with e.g. “long amplicons”, which is more objective.
Line 386: I consider OTU/ASV tables with taxonomy classifications, read abundances per sample, and one representative FASTA as mandatory supplemental item. Please amend as an annotated .csv file.
Thanks!
https://doi.org/10.24072/pci.microbiol.100014.rev12



