
Median review duration = 24 days
Median time to 1st decision = 62 days
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
06 Apr 2025

The cutting type of vegetables influences the spontaneous fermentation rateCutting Type as a Key Factor in shaping Microbial Dynamics during Vegetable FermentationRecommended by Souhir Marsit based on reviews by Thibault Nidelet and Kate HowellFermented vegetables, traditionally consumed in Asian and Eastern countries, are gaining increasing interest in Western countries due to the growing demand for more natural, healthy, and sustainable food. Their potential health effects have only recently begun to be scientifically studied (Thierry et al., 2023). The manufacturing process of fermented vegetables consists of cutting and packing raw vegetables with salt or brine, that will draw water and nutrients out from the vegetable tissue, thus providing microorganisms with the necessary substrates to initiate spontaneous fermentation (Buckenhueskes, 2015). Various parameters, including the cutting method, which may influence the rate of solute diffusion from vegetable tissue, can affect fermentation speed and, consequently, the quality of fermented vegetables. However, the role of cutting type has rarely been addressed. The study by Valence et al. (2025) used a comprehensive range of methods to investigate how cutting types and a slight reduction in salt concentration influence the spontaneous fermentation of two vegetables, carrot and cabbage. Two cutting types, finely or roughly cut, and two salt levels, 1% (the minimum concentration usually used) and a lower salt level in line with health recommendations (0.8%), were tested. Carrot and cabbage fermentations were performed under controlled conditions in duplicate, and microbiological and biochemical characteristics were monitored over one month by combining several approaches and extensive experiments in culturomics, 16S rRNA gene and gyrB metataxonomics for bacterial community analysis, and targeted metabolomics. The study shows the sequential establishment of microbial communities during the fermentation of both vegetables. In the early stages, Enterobacteriaceae replaced the initial microbiota, but they were rapidly outcompeted by Lactic Acid Bacteria (LAB). LAB growth acidified the medium, inhibiting enterobacteria and ensuring microbial safety. Their dominance was attributed to their ability to ferment carbohydrates into lactic acid and possibly the production of antimicrobial compounds. The results of targeted metabolomic analysis show that the main fermentation byproducts are mannitol, lactic acid, and acetic acid, which is consistent with previous studies on fermented vegetables. Most notably, this study demonstrated for the first time that the type of vegetable cutting has a major impact on fermentation dynamics by influencing the release of solutes into the brine. Finer cuts, which provide a greater surface area, facilitate nutrient diffusion, thereby promoting LAB proliferation and acidification. The study also shows that salt addition improved solute release, though the microbial effects were less clear due to variability between replicates. Indeed, significant variability between jars was noted, affecting microbial composition, metabolite profiles, and acidification rates. The work of Valence et al. (2025) highlights for the first time the crucial role of cutting type in vegetable fermentation, demonstrating that finer cuts accelerate acidification, improve microbial safety, and enhance fermentation efficiency. Their findings contribute to the optimization of fermentation processes, providing valuable insights for enhancing the quality of fermented vegetables. References: Thierry A, Baty C, Marché L, Chuat V, Picard O, Lortal S, Valence F. Lactofermentation of vegetables: An ancient method of preservation matching new trends. Trends Food Sci Technol. 2023. https://doi.org/10.1016/j.tifs.2023.07.009 Valence F, Junker R, Baty C, Rué O, Mariadassou M, Madec M, Maillard M, Bage A, Chuat V, Marché L, Thierry A. The cutting type of vegetables influences the spontaneous fermentation rate. HAL, ver.2 2025. https://hal.science/hal-04701063v2 | The cutting type of vegetables influences the spontaneous fermentation rate | Florence Valence, Romane Junker, Céline Baty, Olivier Rué, Mahendra Mariadassou, Marie-Noelle Madec, Marie-Bernadette Maillard, Anne-Sophie Bage, Victoria Chuat, Laurent Marché, Anne Thierry | <p>Fermented vegetables are mainly produced by the spontaneous fermentation of raw vegetables that are roughly or thinly cut, salted and incubated in an oxygen-free environment. Despite the variety of cutting types and their potential role in the ... | | Microbial ecology and environmental microbiology, Microbial physiology, ecophysiology and metabolism, Microbiomes, Molecular microbiology | Souhir Marsit | Kate Howell, Thibault Nidelet | 2024-09-20 17:01:46 | |
21 Jan 2025
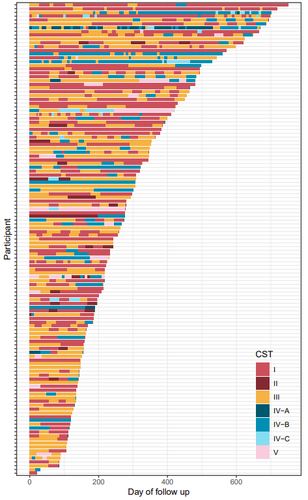
Factors shaping vaginal microbiota long-term community dynamics in young adult womenElucidating microbial community transitions within the human vaginal environmentRecommended by Rafael Muñoz-Tamayo based on reviews by Chen Liao, Simon Labarthe and 1 anonymous reviewer based on reviews by Chen Liao, Simon Labarthe and 1 anonymous reviewer
The human vaginal microbiota plays a key role in urogenital health. Enhancing our understanding of the dynamics of the vaginal microbiota can provide valuable insights for maintaining health and design strategies to prevent urogenital diseases. Health status evolves over time. The work by Kamiya et al. (2024) addressed the dynamic interplay between vaginal microbiota and health using a robust, high-resolution longitudinal cohort of 125 reproductive-aged women, followed for a median duration of 8.6 months in Montpellier, France. The participants were recruited within the PAPCLEAR study, which aimed to better understand the course and natural history of human papillomaviruses infections in healthy, young women (Murall et al. 2019). Each participant contributed at least three vaginal samples, from which microbiota barcoding was performed. The vaginal microbiota was clustered using the approach developed by Ravel et al. (2011) which categorizes microbial communities in 5 community state types with varying health implications. Transitions between community states were estimated using a hierarchical Bayesian Markov model. These transitions were associated with 16 covariates covering lifestyle, sexual practices and medication. This hierarchical approach allowed for the quantification of individual differences among women. The study characterized the stability of vaginal microbial communities and identified alcohol consumption as the strongest covariate driving community transitions. The results indicated that alcohol consumption promotes non-optimal communities. The modelling approach, however, indicated that individual variability among the women was not fully accounted for by the selected 16 covariates, suggesting the need to explore additional key factors, including dynamic covariates. The authors clearly identified several potential limitations of the study, including the variability associated to home sampling, the resolution of the microbial categories, and the impact of the clustering method on the analysis. My decision to recommend this manuscript is supported by the solid and rigorous analysis of the study, strengthened by the clear presentation of methods, data and analysis. While applying advanced computational techniques, the authors provide a solid biological interpretation of their results. This work makes a substantial contribution by expanding the understanding of vaginal microbiota dynamics and its interplay with health. It sets a framework for further evaluation of strategies aimed at promoting vaginal health. Moreover, it presents a generic methodology that could be applied to other microbial ecosystems. References Kamiya T, Tessandier N, Elie B, Bernat C, Boué V, Grasset S, Groc S, Rahmoun M, Selinger C, Humphrys MS, Bonneau M, Graf C, Foulongne V, Reynes J, Tribout V, Segondy M, Boulle N, Ravel J, Murall CL, Alizon S (2024) Factors shaping vaginal microbiota long-term community dynamics in young adult women. medRxiv, 2024.04.08.24305448, ver.3 peer-reviewed and recommended by PCI Microbiol. https://doi.org/10.1101/2024.04.08.24305448 Murall CL, Rahmoun M, Selinger C, Baldellou M, Bernat C, Bonneau M, Boué V, Buisson M, Christophe G, D’Auria G, Taroni F De, Foulongne V, Froissart R, Graf C, Grasset S, Groc S, Hirtz C, Jaussent A, Lajoie J, Lorcy F, Picot E, Picot MC, Ravel J, Reynes J, Rousset T, Seddiki A, Teirlinck M, Tribout V, Tuaillon É, Waterboer T, Jacobs N, Bravo IG, Segondy M, Boulle N, Alizon S (2019) Natural history, dynamics, and ecology of human papillomaviruses in genital infections of young women: protocol of the PAPCLEAR cohort study. BMJ Open, 9, e025129. https://doi.org/10.1136/BMJOPEN-2018-025129 Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, Brotman RM, Davis CC, Ault K, Peralta L, Forney LJ (2011) Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences of the United States of America, 108. https://doi.org/10.1073/pnas.1002611107
| Factors shaping vaginal microbiota long-term community dynamics in young adult women | Tsukushi Kamiya, Nicolas Tessandier, Baptiste Elie, Claire Bernat, Vanina Boue, Sophie Grasset, Soraya Groc, Massilva Rahmoun, Christian Selinger, Michael S. Humphrys, Marine Bonneau, Vincent Foulongne, Christelle Graf, Jacques Reynes, Vincent Tri... | <p>The vaginal microbiota is known to affect women’s health. Yet, there is a notable paucity of high-resolution follow-up studies lasting several months, which would be required to interrogate the long-term dynamics and associations with demograph... | | Mathematical modeling of microbial processes and ecosystems, Microbe-microbe and microbe-host interactions, Microbial ecology and environmental microbiology, Microbiomes | Rafael Muñoz-Tamayo | Simon Labarthe, Anonymous | 2024-09-02 17:27:41 | |
14 Jan 2025

Diel changes in the expression of a marker gene and candidate genes for intracellular amorphous CaCO3 biomineralization in MicrocystisGenetically controlled biomineralization in Cyanobacteria: diel fluctuations of ccyA transcript abundances and identification of neighboring genes putatively involved in the precipitation of intracellular amorphous calcium carbonates in Microcystis aeruginosa PCC7806Recommended by Rutger De Wit based on reviews by Rutger De Wit and 1 anonymous reviewer
In this interesting study by Bruley et al. (2024), the cyanobacterium Microcystis aeruginosa PCC7806 is taken as a model organism for intracellular CaCO3 precipitation in Cyanobacteria, i.e. in the form of intracellular amorphous calcium carbonates (iACC). This phenomenon, which was first described in 2012, is an example of genetically controlled biomineralization in bacteria. Hence, a gene coding for the protein calcyanin (ccyA) has been documented in iACC biomineralizing cyanobacteria. Nevertheless, so far, the functioning of the calcyanin protein remains unknown. As a first step to elucidate its role in iACC biomineralization the authors study the diel variations of ccyA expression. An approximately 2.5-fold variation in abundance of ccyA transcripts has been observed with the highest values of ccyA expression observed during the second half of the dark period. In addition, the authors made a thorough investigation of transcriptomics data, to detect gene-expressions with temporal patterns that positively or negatively correlate with ccyA. A particular focus was made on neighboring genes (both upstream and downstream) to detect a possible operon gathering ccyA with other genes. Very interestingly, the authors discovered that some neighboring genes coding for Ca2+/H+ antiporter systems, showed transcripts with abundances that correlate with that of ccyA. This study raises many interesting questions on genetically controlled biomineralization in bacteria and more particularly the function of iACC biomineralization in Cyanobacteria. As the authors write, iACC biomineralization could be involved in carbon-concentrating mechanisms (CCM), intracellular pH buffering, and create “ballast” for buoyancy and floatability regulation. Nevertheless, these roles would require mechanisms for the dissolution of iAAC in concert with its precipitation ; fine-tuning of both resulting in homeostasis or cyclic temporal patterns of iAAC increase/decrease. As a perspective, the response of Microcystis to fluctuations in calcium and/or pCO2 levels could provide valuable insights into the molecular mechanisms underlying the biomineralization of iACC, as well as comparisons with non-iACC biomineralizing strains or with a mutant of PCC 7806 with a deactivated/deleted ccyA gene. Reference: Bruley A, Gaëtan J, Gugger M, Pancrace C, Millet M, Gaschignard G, Dezi M, Humbert J-F, Leloup J, Skouri-Panet F, Callebaut I, Benzerara K and Duprat E (2024) Diel changes in the expression of a marker gene and candidate genes for intracellular amorphous CaCO3 biomineralization in Microcystis. bioRxiv, ver.3 peer-reviewed and recommended by PCI Microbiol. https://doi.org/10.1101/2024.07.07.602159
| Diel changes in the expression of a marker gene and candidate genes for intracellular amorphous CaCO3 biomineralization in *Microcystis* | Apolline Bruley, Juliette Gaëtan, Muriel Gugger, Claire Pancrace, Maxime Millet, Geoffroy Gaschignard, Manuela Dezi, Jean-François Humbert, Julie Leloup, Fériel Skouri-Panet, Isabelle Callebaut, Karim Benzerara, Elodie Duprat | <p>Phylogenetically diverse cyanobacteria biomineralize intracellular amorphous calcium carbonate (iACC) inclusions. This includes several genotypes of the Microcystis genus, a potentially toxic, bloom-forming cyanobacterium found worldwide in fre... | | Microbial biogeochemistry, Microbial ecology and environmental microbiology, Microbial physiology, ecophysiology and metabolism | Rutger De Wit | 2024-07-11 17:56:28 | ||
28 Nov 2024
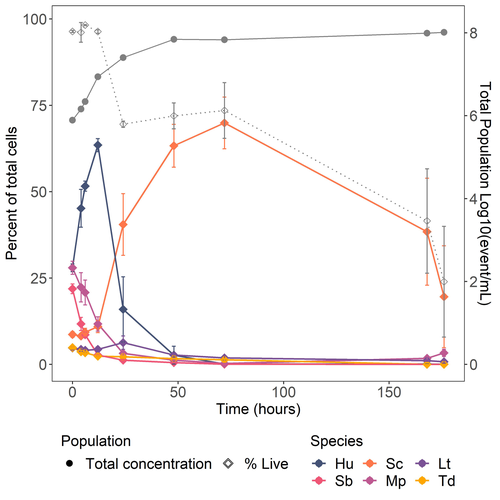
Design of a new model yeast consortium for ecological studies of enological fermentationYeast consortium for novel wine fermentationsRecommended by Francisco Cubillos based on reviews by Pablo Villarreal, Cristian Varela and 3 anonymous reviewersThe article by Pourcelot et al. (2024) brings a novel approach to wine fermentation. Recently, scientific advances have focused on utilizing microbial consortiums rather than individual species alone or even two individuals co-inoculated. However, spontaneous fermentations are complex, and microbes work in communities. This work aims to design a yeast consortium by studying the population changes over time and determining the metabolite production and fermentation kinetics. In this way, the authors present an elegant molecular approach by tagging each strain to construct a wine fermentation consortium. References Eléonore Pourcelot, Audrey Vigna, Thérèse Marlin, Virginie Galeote, Thibault Nidelet (2024) Design of a new model yeast consortium for ecological studies of enological fermentation. bioRxiv, ver.4 peer-reviewed and recommended by PCI Microbiol https://doi.org/10.1101/2024.05.06.592697 | Design of a new model yeast consortium for ecological studies of enological fermentation | Eléonore Pourcelot, Audrey Vigna, Thérèse Marlin, Virginie Galeote, Thibault Nidelet | <p>Wine fermentation involves complex microbial communities of non-<em>Saccharomyces</em> yeast species besides the well-known <em>Saccharomyces cerevisiae</em>. While extensive research has enhanced our understanding of <em>S. cerevisiae</em>, th... | | Microbial ecology and environmental microbiology | Francisco Cubillos | Cristian Varela, Pablo Villarreal, Anonymous | 2024-05-24 12:17:23 | |
21 Nov 2024
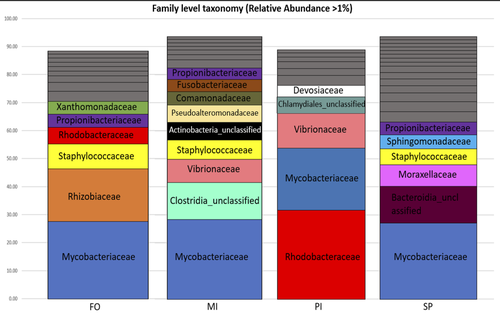
The effect of dietary fish oil replacement by microalgae on the gilthead sea bream midgut bacterial microbiotaInsights on the gilthead sea bream midgut microbiota adaptation to three types of microalgal-based dietsRecommended by Angélique Gobet based on reviews by Yaqiu Liu and 1 anonymous reviewer
In fed aquaculture, fishes are commonly fed with a fish-oil based diet mostly coming from captured fishes. This is one main global issue leading to overfishing of wild species (Cashion et al., 2017; Tacon & Metian, 2008). Several alternatives in lipid sources for fish diet have been tested and promising alternatives such as plants (e.g. rapeseed oil) or microalgae (e.g. Schizochytrium sp.) have been identified (Pérez-Pascual et al., 2020). Like other animals, fishes’ digestive tract is composed of a microbiota whose composition is linked to the host physiological state as well as its diet (Yukgehnaish et al., 2020). In reared fishes such as the European sea bass (Dicentrarchus labrax), replacing fish oil by other sources such as microalgae in their diet has been shown to modify the digestive microbiota composition (Pérez-Pascual et al., 2020). Here, the aim of Katsoulis-Dimitriou et al. (2024), was to test the effect of three dietary microalgae blends on the midgut microbiota composition of the reared fishes. The authors compared the effect of a control diet (i.e. with only fish oil as lipid source, namely, FO) with that of three experimental diets with two thirds of the fish oil replaced by either a mixture of the microalgae Microchloropsis gaditana and Isochrysis sp. (now known as Tisochrysis lutea, MI), Phaeodactylum tricornutum and Isochrysis sp. (PI) or Schizochytrium sp. and P. tricornutum (SP). For each diet, 25 fishes were reared in each of the triplicated tanks and, after 80 days of experiment, a total of 10 fishes per diet were sampled. DNA was extracted from the midgut part of the intestine and a 16S rDNA-based metabarcoding approach was conducted to survey the associated bacterial community. Each diet type, FO, MI, PI and SP, was mostly characterized by a composition of specific abundant OTUs, indicating the clear influence of the oil composition on the digestive microbiota. When feeding with the MI diet, the authors also highlighted the presence of some candidate genera (e.g. Pseudoalteromonas, Pseudomonas, Bacillus and Rhodopseudomonas) as potential probiotics for fish aquaculture. Finally, in comparison to the fish oil diet, a predictive metabolic analysis of the bacterial community could suggest a differential expression of some polysaccharide metabolisms with the microalgae-based diets, highlighting a probable diet-based effect on the microbiota functioning. The work from Katsoulis-Dimitriou et al. (2024) completes the current knowledge on using sustainable alternatives to traditional fish feed and its effect on the digestive microbiota composition of fishes. This work also opens new ways to be explored considering the enrichment of potential probiotics using microalgae-base diets. Further analyses testing specific functional approaches (e.g. transcriptomics, metabolomics) may allow completing the understanding of the gut microbiota functioning linked to diet composition. Finally, measurements on fish biometrics in a similar experiment should help understanding the contribution of a microalgal-diet to fish fitness. References Cashion, T., Le Manach, F., Zeller, D., & Pauly, D. (2017). Most fish destined for fishmeal production are food‐grade fish. Fish and Fisheries, 18(5), 837–844. https://doi.org/10.1111/faf.12209 Katsoulis-Dimitriou, S., Nikouli, E., Gkalogianni, E., Karapanagiotidis, I., Kormas, K. (2024) The effect of dietary fish oil replacement by microalgae on the gilthead sea bream midgut bacterial microbiota. BioRxiv, ver.3 peer-reviewed and recommended by PCI Microbiol https://doi.org/10.1101/2024.01.24.576938 Pérez-Pascual, D., Estellé, J., Dutto, G., Rodde, C., Bernardet, J.-F., Marchand, Y., Duchaud, E., Przybyla, C., & Ghigo, J.-M. (2020). Growth Performance and Adaptability of European Sea Bass (Dicentrarchus labrax) Gut Microbiota to Alternative Diets Free of Fish Products. Microorganisms, 8(9), 1346. https://doi.org/10.3390/microorganisms8091346 Tacon, A. G. J., & Metian, M. (2008). Global overview on the use of fish meal and fish oil in industrially compounded aquafeeds: Trends and future prospects. Aquaculture, 285(1–4), 146–158. https://doi.org/10.1016/j.aquaculture.2008.08.015 Yukgehnaish, K., Kumar, P., Sivachandran, P., Marimuthu, K., Arshad, A., Paray, B. A., & Arockiaraj, J. (2020). Gut microbiota metagenomics in aquaculture: Factors influencing gut microbiome and its physiological role in fish. Reviews in Aquaculture, 12(3), 1903–1927. https://doi.org/10.1111/raq.12416
| The effect of dietary fish oil replacement by microalgae on the gilthead sea bream midgut bacterial microbiota | Stefanos Katsoulis-Dimitriou, Eleni Nikouli, Elli-Zafeiria Gkalogianni, Ioannis Karapanagiotidis, Konstantinos Kormas | <p> It is well known that the gut microbiome and its interaction with the host influence several important factors for fish health such as nutrition and metabolism. Diet is one of the main factors influencing the composition of the gut microb... | | Microbe-microbe and microbe-host interactions, Microbial symbiosis, Microbiomes | Angélique Gobet | 2024-01-25 18:09:56 | ||
19 Jul 2024

Microbiome turnover during offspring development varies with maternal care, but not moult, in a hemimetabolous insectStability in a microbe-insect interactionRecommended by Konstantinos Kormas based on reviews by Guillame Minard and Enric Frago
The degree of fidelity between microbes and their hosts varies considerably among different animal groups but also along the host's developmental stages and depends on the stability of their microbial communities. Cheutin et al. showcase experimentally the stability of whole body bacterial microbiome in a dermapteran insect species, the European earwig Forficula auricularia. The carefully designed experiments, which include a large number of investigated families and the related methodologies along with the data analysis, revealed that the bacterial communities of this insect are highly dynamic during the early developmental stages, but these changes are rather specific to each developmental stage and rather irrelevant to moulting. Some of these changes were reflected in the dominant predicted metabolic pathways. Another important finding of this study was that maternal care of the eggs has a detectable impact on the future shaping of the adult insect bacterial microbiome. The findings of this paper clearly answer its working hypotheses, but they also generate a set of specific novel hypotheses for future studies. These hypotheses are of interest to the general field of animal-microbe interactions and, more specifically, to the driving forces of transmissability of microbes from one generation to the next one. This study also depicts some of the most likely important metabolic pathways in this insect-microbe relationship that could be the focus of future studies with more specific methodologies. References Cheutin M-C, Boucicot M, Meunier J. (2024). Microbiome turnover during offspring development varies with maternal care, but not moult, in a hemimetabolous insect. bioRxiv, ver.3, peer-reviewed and recommended by Peer Community In Microbiology. https://www.biorxiv.org/content/10.1101/2024.03.26.586808v3 | Microbiome turnover during offspring development varies with maternal care, but not moult, in a hemimetabolous insect | Marie-Charlotte Cheutin, Manon Boucicot, Joel Meunier | <p>The ecological success of insects often depends on their association with beneficial microbes. However, insect development involves repeated moults, which can have dramatic effects on their microbial communities. Here, we investigated whether a... | | Microbial ecology and environmental microbiology, Microbial physiology, ecophysiology and metabolism, Microbiomes | Konstantinos Kormas | 2024-03-28 12:24:50 | ||
29 May 2024
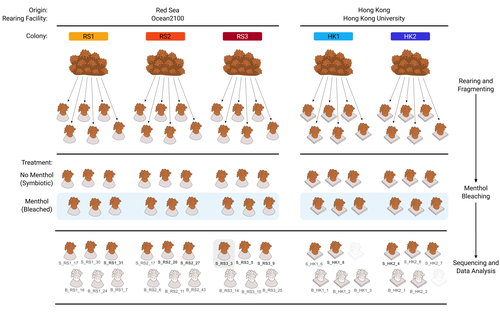
The bacterial microbiome of symbiotic and menthol-bleached polyps of long-term aquarium-reared Galaxea fascicularisAn important step forward in deciphering coral symbiosis through manipulative approachesRecommended by Yui Sato based on reviews by Tony Robinet and 1 anonymous reviewer
As complex multipartite interactions among the coral host and coral-associated microbial entities including the dinoflagellate symbionts, bacteria, archaea and viruses, have been appreciated, a manipulatable, less-complex study system is desired to deepen our functional understanding of this fascinating symbiotic system. Among experimental manipulation approaches, removal of the algal symbionts using menthol is widely implemented; however, its effect on the rest of the coral-associated symbiotic members has not been explored, which is critical knowledge to assess experimental works using this popular method. This preprint by Puntin et al. (https://doi.org/10.1101/2023.08.23.554380) presents an important observation in this aspect. Their initial observations suggest that menthol-induced coral bleaching introduces stochastic changes in associated bacterial communities, which resemble dysbiosis, making bacterial communities more dissimilar from each other. They also observed low taxonomic diversity in bacterial communities on the corals maintained in aquaria over several months, worth noting as a positive value as an experimental system. Their data are preliminary by nature, while they present intriguing ideas that warrant further studies. Reference Puntin G, Wong JCY, Röthig T, Baker DM, Sweet M, Ziegler M (2024). The bacterial microbiome of symbiotic and menthol-bleached polyps of long-term aquarium-reared Galaxea fascicularis (2024). bioRxiv, ver.4., peer-reviewed and recommended by Peer Community In Microbiology. https://doi.org/10.1101/2023.08.23.554380
| The bacterial microbiome of symbiotic and menthol-bleached polyps of long-term aquarium-reared *Galaxea fascicularis* | Giulia Puntin, Jane C.Y. Wong, Till Roethig, David M. Baker, Michael Sweet, Maren Ziegler | <p>Coral reefs support the livelihood of half a billion people but are at high risk of collapse due to the vulnerability of corals to climate change and local anthropogenic stressors. While understanding coral functioning is essential to guide con... | | Microbial symbiosis, Microbiomes | Yui Sato | 2023-08-26 04:50:01 | ||
10 May 2024
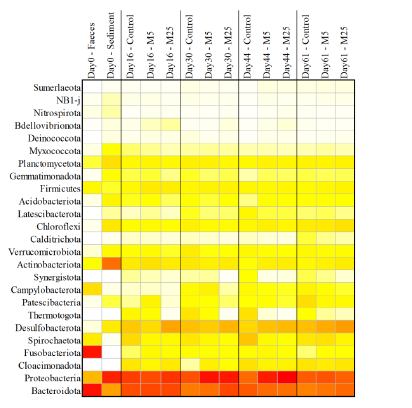
Molybdate delays sulphide formation in the sediment and transfer to the bulk liquid in a model shrimp pondAddition of molybdate to shrimp ponds is a promising new technique to delay the accumulation of toxic H2SRecommended by Roey Angel based on reviews by 2 anonymous reviewers
Shrimp aquaculture ponds are an established technology that helps answer the demand for high-protein food while reducing the impact of fishing on the oceans. However, as a closed system, high in organic matter, aquaculture ponds in general and those used for shrimp in particular tend to develop anoxic sediments and favour sulfate reduction to H2S. The development of hydrogen sulphide, in return, is toxic to the shrimp and can lead to lower yields. A standard solution to the problem is to inject air into the sediments. However, this solution requires additional infrastructure, is costly to operate, and can also disturb other essential life forms in the pond, such as benthic plants. In this work by Torun et al. (2024), the authors used a carefully designed lab model of shrimp ponds to show that the addition of molybdate at concentrations as low as 5 mg/l delayed the accumulation of H2S and pushed the zone rich in sulphide deeper into the sediment. The postulated mechanism for the inhibition in H2S production is that molybdate binds to the ATP sulfurylase in sulphate-reducing bacteria (SRB), and together with ATP, they generate adenosine 5′-phosphosulfate (APS) that cannot be used as an electron acceptor. Surprisingly, however, the growth of SRB was stimulated rather than inhibited in this experiment. While the exact cause remains unknown, the authors postulate that SRB resorted to alternative metabolic pathways such as fermentation. Overall, while this work was done on a model system in the lab, adding molybdate to shrimp aquaculture ponds is a promising technique and should be tested on a larger scale. Reference Torun F, Hostins B, Schryver PD, Boon N, Vrieze JD. (2024). Molybdate delays sulphide formation in the sediment and transfer to the bulk liquid in a model shrimp pond. bioRxiv, ver.3, peer-reviewed and recommended by Peer Community In Microbiology. https://doi.org/10.1101/2023.11.16.567380 | Molybdate delays sulphide formation in the sediment and transfer to the bulk liquid in a model shrimp pond | Funda Torun, Barbara Hostins, Peter De Schryver, Nico Boon, Jo De Vrieze | <p>Shrimp are commonly cultured in earthen aquaculture ponds where organic-rich uneaten feed and faeces accumulate on and in the sediment to form anaerobic zones. Since the pond water is rich in sulphate, these anaerobic conditions eventually lead... | | Microbial biotechnology, Microbial ecology and environmental microbiology, Microbiomes | Roey Angel | 2023-11-20 12:08:51 | ||
12 Apr 2024
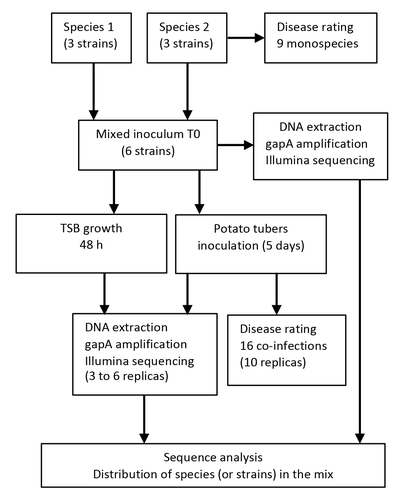
Bacterial pathogens dynamic during multi-species infectionsUnraveling disease ecology: insights from soft rot Pectobacteriaceae co-infectionsRecommended by Clara Torres-Barceló based on reviews by 2 anonymous reviewersFew studies deal with the understanding of disease ecology, especially in the agricultural domain. Soft rot Pectobacteriaceae are major plant pathogens that frequently co-infect potato tubers. Exploring their ecological relationships can provide valuable insights for effective monitoring and preventing disease. The study of Barny et al (2024) explores the dynamics of synthetic communities of soft rot Pectobacterium species (SRP) following in vitro and in vivo inoculations, focusing on the implications for disease development. To delve into co-infection dynamics, the authors constructed mixed populations comprising six strains, with three strains from each of two species. Through inoculations of both liquid cultures and potatoes, they observed outcomes using amplicon sequencing targeting the gapA gene, along with monitoring bacterial population sizes and symptoms on potato tubers. Results reveal intriguing patterns: competition among strains of the same species, cooperation through trophic interactions, and interference due to toxicity. Thanks to a modelling approach, they suggest that the presence of a cheater strain may be favoured when it is associated with an aggressive strain. This finding is crucial for field sampling strategies, as there is a risk that during an outbreak, only the cheater strain may be detected, potentially overlooking the problematic aggressive strain. While the study conducted by Barny et al. (2024) provides valuable insights into strain interactions, it also highlights areas for further exploration to enhance understanding. First, the extent to which different species occupy similar niches in real agricultural scenarios remains unclear. Additionally, comparative genomics analysis on strains and investigating specific gene candidates could offer valuable mechanistic insights into strain dynamics. These areas for future research offer chances to build up our knowledge base in this field and improve how we understand the interactions between bacteria in nature. The implications of the study extend beyond plant pathogens like SRP. Similar scenarios of complex diseases involving closely related species or strains competing within the same niche are observed in human pathogens as well. Reference Barny, M.-A., Thieffry, S., Gomes de Faria, C., Thebault, E., Pedron, J. (2024). Bacterial pathogens dynamic during multi-species infections. https://doi.org/10.1101/2023.12.06.570389
| Bacterial pathogens dynamic during multi-species infections | Marie-Anne Barny, Sylvia Thieffry, Christelle Gomes de Faria, Elisa Thebault, Jacques Pedron | <p>Soft rot Pectobacteriacea (SRP) gathers more than 30 bacterial species that collectively rot a wide range of plants by producing and secreting a large set of plant cell wall degrading enzymes (PCWDEs). Worldwide potato field surveys identified ... | | Microbe-microbe and microbe-host interactions, Microbial ecology and environmental microbiology | Clara Torres-Barceló | 2023-12-12 17:54:07 | ||
04 Jan 2024
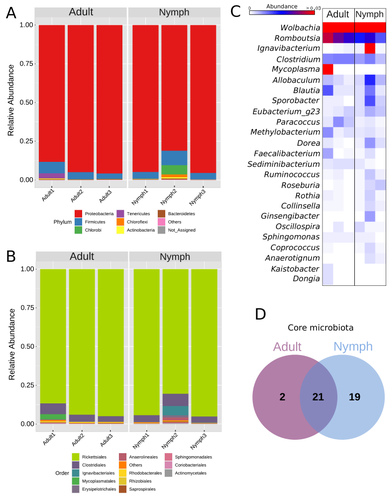
Diversity of bacterial symbionts associated with the tropical plant bug Monalonion velezangeli (Hemiptera: Miridae) revealed by high-throughput 16S-rRNA sequencingShedding light on bacteria associated with an agricultural pest, the tropical plant bug Monalonion velezangeli: a foundational descriptive studyRecommended by Jean-Marie Volland based on reviews by 2 anonymous reviewers
The paper "Diversity of bacterial symbionts associated with the tropical plant bug Monalonion velezangeli (Hemiptera: Miridae) revealed by high-throughput 16S rRNA sequencing" by Navarro-Escalante et al. (2023) is a valuable contribution to entomological research, particularly in the context of pest management. This descriptive study, while not delving into the functional characterization of the associated bacterial strains, lays an essential groundwork for understanding the bacterial components of the microbiota of this agricultural pest. This study is interesting because it provides new information on insect microbiota, especially in a family for which the knowledge of the diversity of bacterial symbionts is very limited. One of the study's core strengths lies in its exploration and definition of the core microbiota of M. velezangeli, which could serve as a foundation for future research aimed at pest control strategies. The use of 16S rRNA sequencing, despite its known limitations, has enabled the profiling of these bacterial communities. The paper highlights the absence of differences in the bacterial communities associated with the nymph and adult stages of the pest, indicating a stable association of these microbes throughout the insect's life cycle. A standout point in the study is the overwhelming presence of the symbiont Wolbachia, accounting for approximately 92% of the bacterial composition. However, intriguingly, the authors also note the absence of Wolbachia in some individuals, suggesting a more complex dynamic that warrants further investigation. This finding is particularly noteworthy, as it opens up questions about the role of Wolbachia and its impact on the biology and ecology of M. velezangeli. The researchers have carefully addressed all the reviewers’ comments and suggestions. They also addressed a potential bias in their study - the overwhelming presence of Wolbachia - by analyzing the bacterial community after the removal of Wolbachia sequences. This careful approach enriches the study's credibility and ensures a more accurate representation of the pest's microbiota. The identification of potentially culturable strains within the core microbiome represents an interesting perspective of this research. This information could be used in future efforts to develop pest control strategies, particularly those employing paratransgenic approaches. The possibility of manipulating these culturable strains to combat M. velezangeli presents an exciting avenue for sustainable pest management. While the study does not investigate the localization of these associated bacteria, whether in the gut or elsewhere, including potentially in dedicated symbiotic organs, it nevertheless offers a valuable descriptive account. This baseline knowledge will be useful for any subsequent functional or localization studies, which could further unravel the complex interactions between M. velezangeli and its microbial partners. In conclusion, the work of Navarro-Escalante et al. is a notable effort to set the stage for future research into the biology of M. velezangeli and its associated microbiota. The findings from this study provide a good reference point for further investigations aimed at pest's biology and exploring innovative pest control strategies. It also represents a valuable contribution to understanding the basic biology of insect-bacteria interactions.
| Diversity of bacterial symbionts associated with the tropical plant bug *Monalonion velezangeli* (Hemiptera: Miridae) revealed by high-throughput 16S-rRNA sequencing | Lucio Navarro-Escalante, Pablo Benavides, Flor Edith Acevedo | <p>Insects and microbes have developed complex symbiotic relationships that evolutionarily and ecologically play beneficial roles for both, the symbiont and the host. In most Hemiptera insects, bacterial symbionts offer mainly nutritional, defensi... | | Microbial ecology and environmental microbiology, Microbial symbiosis | Jean-Marie Volland | 2022-10-31 20:31:54 |




