
Average time to find at least 2 reviewers after submission = 26 days (median = 17)
Average time from submission to 1st decision = 68 days (median = 57)
Latest recommendations

| Id | Title * | Authors * ▼ | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
09 May 2023
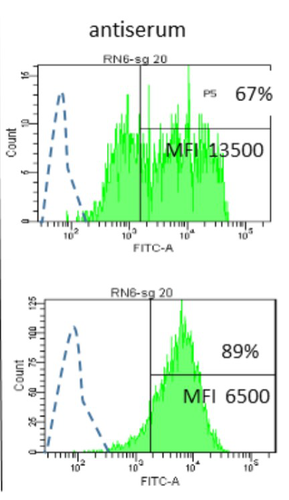
Interactions between Mycoplasma mycoides subsp. mycoides and bovine macrophages under physiological conditionsInteraction of bovine macrophages with Mycoplasma mycoides subsp. mycoidesRecommended by Pablo Zunino based on reviews by 2 anonymous reviewersMycoplasma mycoides subsp. mycoides (Mmm), a pathogenic wall-less bacterium, is the etiological agent of contagious bovine pleuropneumonia (CBPP). This highly contagious respiratory disease may develop in severe pneumonia, with associated high mortality rates in cattle. Mmm can display different immune evasion mechanisms; in addition, a host uncontrolled inflammatory response stands for lung lesions and chronic carrier animals. Macrophages are among the most important lines of defense against Mmm of the lower respiratory tract. Although their importance in defense and immune response modulation is known, results about their role and mechanisms of action are scarce and sometimes conflicting. In the present study, Totté et al. (1) aimed to investigate the interaction of bovine macrophages (isolated from cattle peripheral blood mononuclear cells) with Mmm, under in vitro conditions. The authors highlight that the study was performed under physiological conditions (in the presence of complement prepared from the same cell donor). In their study, using different approaches, the authors provide interesting and original results, proposing a pivotal role of complement in controlling the inflammatory response, which is crucial in the CBPP pathogenesis. The authors reported that macrophages did not kill Mmm in the presence of a non-bactericidal concentration of bovine serum. However, Mmm inactivation was observed when antiserum from CBPP convalescent animals was used. They also observed that Mmm induced the production of TNF by macrophages (when a high MOI was assessed). However, complement could even abolish Mmm-induced TNF response when used at bactericidal activity concentrations. This role of complement could be combined with the development of potentially protective antibodies against particular Mmm antigens involved in the interaction with identified macrophage receptors to propose control strategies against CBPP. Overall, the study by Totté et al. provides new fundamental insight for the research on preventive or therapeutic strategies for a poorly understood disease that still represents a serious concern for livestock production. REFERENCES 1. Totté, P., Bonnefois, T., Manso-Silván, L. Interactions between Mycoplasma mycoides subsp. mycoides and bovine macrophages under physiological conditions. bioRxiv 2022.12.06.519279, ver. 2 peer-reviewed and recommended by Peer Community In Microbiology. https://doi.org/10.1101/2022.12.06.519279 | Interactions between *Mycoplasma mycoides* subsp. *mycoides* and bovine macrophages under physiological conditions | Philippe Totté, Tiffany Bonnefois, Lucia Manso-Silvan | <p style="text-align: justify;">Abstract</p> <p>We investigated the interactions of unopsonized and opsonized *Mycoplasma mycoides* subsp. *mycoides* (Mmm) with bovine macrophages *in vitro*. Mmm survived and proliferated extracellularly on bovin... | | Microbe-microbe and microbe-host interactions | Pablo Zunino | Anonymous, Anonymous | 2022-12-09 15:12:53 | |
29 Aug 2023
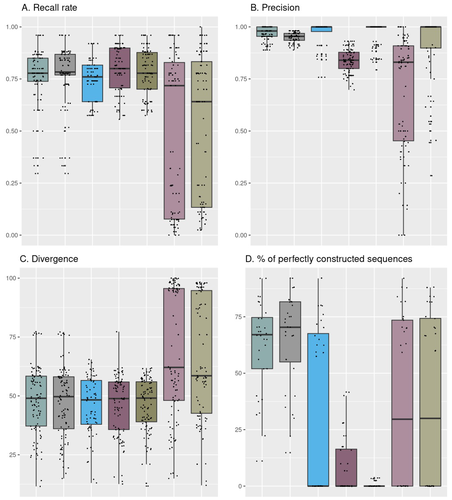
Comparison of metabarcoding taxonomic markers to describe fungal communities in fermented foodsTowards a more accurate metabarcoding approach for studying fungal communities of fermented foodsRecommended by Caroline Strub based on reviews by Johannes Schweichhart and 2 anonymous reviewersImproved characterization of food microbial ecosystems, especially those fermented is key to the development of food sustainability. Short-read metabarcoding is one of the most popular ways to study microbial communities. However, this approach remains complex because of the locks and biases it may entail particularly when applied to fungal communities. Building and using four mock communities from fermented food (bread, wine, cheese, fermented meat), Rué et al., 2023 demonstrate that combined DADA2 denoising algorithm followed to the FROGS tools gives a more accurate description of fungal communities compared to several commonly used bioinformatic workflows, dealing with all amplicon lengths. Moreover, Rué et al., 2023 provide guidance on which barcode to use (ITS1, ITS2, D1/D2 and RPB2), depending on the fermented food studied. Practices in metabarcoding of fungi have been recently reviewed by Tedersoo et al., 2022 and their synthesis comes to the same conclusion as Rué et al., 2023. As the reference databases are far from being complete notably for food ecosystems, the development of specific sequences public databases will enable the scientific community to lift the veil on this whole area of microbial ecology. The study conducted by Rué et al. (2023) provides a particularly detailed approach from a technical point of view, which contributes to improving the general practices in the metabarcoding of fungi. The design and the use of mock communities to compare the performances of the different pipelines is a strong point of this study. Another key element is the creation and use of an in-house database of fungal barcode sequences which improved the species-level affiliations However, the study of fungal communities by metabarcoding is still a promising avenue of research in agri-food sciences. Thus, short-read sequencing, combined with suitable pipelines and databases, should remain of interest to the microbial ecology community (Pauvert et al., 2019; Furneaux et al., 2021). References Furneaux, B., Bahram, M., Rosling, A., Yorou, N. S., & Ryberg, M. (2021). Long‐and short‐read metabarcoding technologies reveal similar spatiotemporal structures in fungal communities. Molecular Ecology Resources, 21(6), 1833-1849. https://doi.org/10.1111/1755-0998.13387 Pauvert, C., Buée, M., Laval, V., Edel-Hermann, V., Fauchery, L., Gautier, A., ... & Vacher, C. (2019). Bioinformatics matters: The accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline. Fungal Ecology, 41, 23-33. https://doi.org/10.1016/j.funeco.2019.03.005 Rué, O., Coton, M., Dugat-Bony, E., Howell, K., Irlinger, F., Legras, J. L., ... & Sicard, D. (2023). Comparison of metabarcoding taxonomic markers to describe fungal communities in fermented foods. BioRxiv, 2023-0113.523754, ver.3 peer-reviewed and recommended by Peer Community in Microbiology. https://doi.org/10.1101/2023.01.13.523754 Tedersoo, L., Bahram, M., Zinger, L., Nilsson, R. H., Kennedy, P. G., Yang, T., ... & Mikryukov, V. (2022). Best practices in metabarcoding of fungi: From experimental design to results. Molecular ecology, 31(10), 2769-2795. https://doi.org/10.1111/mec.16460 | Comparison of metabarcoding taxonomic markers to describe fungal communities in fermented foods | Olivier Rué, Monika Coton, Eric Dugat-Bony, Kate Howell, Françoise Irlinger, Jean-Luc Legras, Valentin Loux, Elisa Michel, Jérôme Mounier, Cécile Neuvéglise, Delphine Sicard | <p>Next generation sequencing offers several ways to study microbial communities. For agri-food sciences, identifying species in diverse food ecosystems is key for both food sustainability and food security. The aim of this study was to compare me... | | Bioinformatics dedicated to microbial studies | Caroline Strub | 2023-01-20 12:37:03 | ||
20 Sep 2023
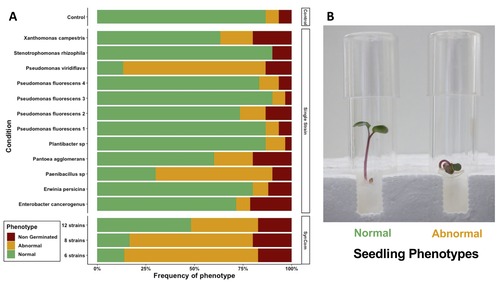
Transmission of synthetic seed bacterial communities to radish seedlings: impact on microbiota assembly and plant phenotypeSeed synthetic community matters and its impact on seedling is strain- and not species-dependantRecommended by Sebastien Massart based on reviews by Cindy Morris, Sebastian Pfeilmeier and 1 anonymous reviewerEngineering plant microbiota can improve plant health and growth sustainably. Emergent approaches include rational Synthetic Communities (SynCom) design or soil amendments and specific agricultural practices to shift resident microbiota and to understand its impact (Moreira et al. 2023). In this context, the impact of seed microbiota on the early stages of plant development is becoming an essential topic in the study of plant–microbiota interactions. Behind the well-studied seed-borne pathogens, the seed microbiota can host many other commensal and beneficial organisms that have been neglected in the past. The study of Simonin et al. (2023) applies single isolates and synthetic communities (SynCom) on radish seeds to answer two key questions: what is the role of seed microbiota during the early stages of plant development? How can SynCom influence the seedling health and its microbiota? The study describes an elegant approach to cope with the variability of natural microbiota using SynCom following a gradient of complexity. Overall, the study highlighted a contrasted impact of the bacterial strains when applied in isolation or SynCom. The composition and complexity of the SynCom had also an impact on plant seedlings. Importantly, contrasting evolution from seeds to seedlings was observed for 3 strains of Pseudomonas fluorescens within the SynComs, underlining the importance of intra-species level diversity and precluding any generalization of results at species level. References Moreira, Z. P. M., Chen, M. Y., Ortuno, D. L. Y., & Haney, C. H. (2023). Engineering plant microbiomes by integrating eco-evolutionary principles into current strategies. Current Opinion in Plant Biology, 71, 102316. https://doi.org/10.1016/j.pbi.2022.102316 Simonin, M., Préveaux, A., Marais, C., Garin, T., Arnault, G., Sarniguet, A., & Barret, M. (2023). Transmission of synthetic seed bacterial communities to radish seedlings: impact on microbiota assembly and plant phenotype. bioRxiv, 2023-02. ver. 3 peer-reviewed and recommended by Peer Community in Microbiology. https://doi.org/10.1101/2023.02.14.527860 | Transmission of synthetic seed bacterial communities to radish seedlings: impact on microbiota assembly and plant phenotype | Marie Simonin, Anne Preveaux, Coralie Marais, Tiffany Garin, Gontran Arnault, Alain Sarniguet, Matthieu Barret | <p style="text-align: justify;">Seed-borne microorganisms can be pioneer taxa during germination and seedling emergence. Still, the identity and phenotypic effects of these taxa that constitute a primary inoculum of plant microbiota is mostly unkn... | | Microbe-microbe and microbe-host interactions, Microbial ecology and environmental microbiology, Microbiomes | Sebastien Massart | 2023-02-15 10:27:26 | ||
19 Jul 2024
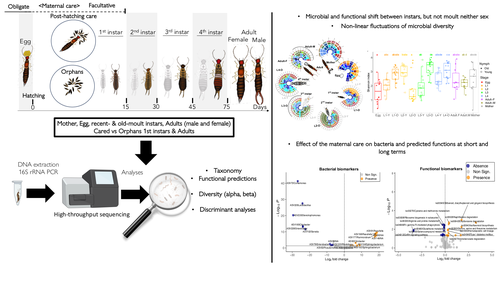
Microbiome turnover during offspring development varies with maternal care, but not moult, in a hemimetabolous insectStability in a microbe-insect interactionRecommended by Konstantinos (Kostas) Kormas based on reviews by Guillame Minard and Enric Frago based on reviews by Guillame Minard and Enric Frago
The degree of fidelity between microbes and their hosts varies considerably among different animal groups but also along the host's developmental stages and depends on the stability of their microbial communities. Cheutin et al. showcase experimentally the stability of whole body bacterial microbiome in a dermapteran insect species, the European earwig Forficula auricularia. The carefully designed experiments, which include a large number of investigated families and the related methodologies along with the data analysis, revealed that the bacterial communities of this insect are highly dynamic during the early developmental stages, but these changes are rather specific to each developmental stage and rather irrelevant to moulting. Some of these changes were reflected in the dominant predicted metabolic pathways. Another important finding of this study was that maternal care of the eggs has a detectable impact on the future shaping of the adult insect bacterial microbiome. The findings of this paper clearly answer its working hypotheses, but they also generate a set of specific novel hypotheses for future studies. These hypotheses are of interest to the general field of animal-microbe interactions and, more specifically, to the driving forces of transmissability of microbes from one generation to the next one. This study also depicts some of the most likely important metabolic pathways in this insect-microbe relationship that could be the focus of future studies with more specific methodologies. References Cheutin M-C, Boucicot M, Meunier J. (2024). Microbiome turnover during offspring development varies with maternal care, but not moult, in a hemimetabolous insect. bioRxiv, ver.3, peer-reviewed and recommended by Peer Community In Microbiology. https://www.biorxiv.org/content/10.1101/2024.03.26.586808v3 | Microbiome turnover during offspring development varies with maternal care, but not moult, in a hemimetabolous insect | Marie-Charlotte Cheutin, Manon Boucicot, Joel Meunier | <p>The ecological success of insects often depends on their association with beneficial microbes. However, insect development involves repeated moults, which can have dramatic effects on their microbial communities. Here, we investigated whether a... | | Microbial ecology and environmental microbiology, Microbial physiology, ecophysiology and metabolism, Microbiomes | Konstantinos (Kostas) Kormas | 2024-03-28 12:24:50 | ||
12 Apr 2024
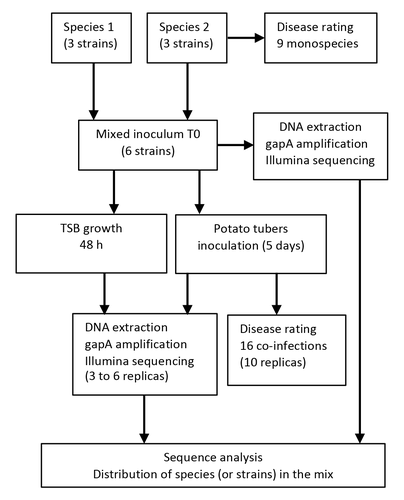
Bacterial pathogens dynamic during multi-species infectionsUnraveling disease ecology: insights from soft rot Pectobacteriaceae co-infectionsRecommended by Clara Torres-Barceló based on reviews by 2 anonymous reviewersFew studies deal with the understanding of disease ecology, especially in the agricultural domain. Soft rot Pectobacteriaceae are major plant pathogens that frequently co-infect potato tubers. Exploring their ecological relationships can provide valuable insights for effective monitoring and preventing disease. The study of Barny et al (2024) explores the dynamics of synthetic communities of soft rot Pectobacterium species (SRP) following in vitro and in vivo inoculations, focusing on the implications for disease development. To delve into co-infection dynamics, the authors constructed mixed populations comprising six strains, with three strains from each of two species. Through inoculations of both liquid cultures and potatoes, they observed outcomes using amplicon sequencing targeting the gapA gene, along with monitoring bacterial population sizes and symptoms on potato tubers. Results reveal intriguing patterns: competition among strains of the same species, cooperation through trophic interactions, and interference due to toxicity. Thanks to a modelling approach, they suggest that the presence of a cheater strain may be favoured when it is associated with an aggressive strain. This finding is crucial for field sampling strategies, as there is a risk that during an outbreak, only the cheater strain may be detected, potentially overlooking the problematic aggressive strain. While the study conducted by Barny et al. (2024) provides valuable insights into strain interactions, it also highlights areas for further exploration to enhance understanding. First, the extent to which different species occupy similar niches in real agricultural scenarios remains unclear. Additionally, comparative genomics analysis on strains and investigating specific gene candidates could offer valuable mechanistic insights into strain dynamics. These areas for future research offer chances to build up our knowledge base in this field and improve how we understand the interactions between bacteria in nature. The implications of the study extend beyond plant pathogens like SRP. Similar scenarios of complex diseases involving closely related species or strains competing within the same niche are observed in human pathogens as well. Reference Barny, M.-A., Thieffry, S., Gomes de Faria, C., Thebault, E., Pedron, J. (2024). Bacterial pathogens dynamic during multi-species infections. https://doi.org/10.1101/2023.12.06.570389
| Bacterial pathogens dynamic during multi-species infections | Marie-Anne Barny, Sylvia Thieffry, Christelle Gomes de Faria, Elisa Thebault, Jacques Pedron | <p>Soft rot Pectobacteriacea (SRP) gathers more than 30 bacterial species that collectively rot a wide range of plants by producing and secreting a large set of plant cell wall degrading enzymes (PCWDEs). Worldwide potato field surveys identified ... | | Microbe-microbe and microbe-host interactions, Microbial ecology and environmental microbiology | Clara Torres-Barceló | 2023-12-12 17:54:07 | ||
04 Jan 2024
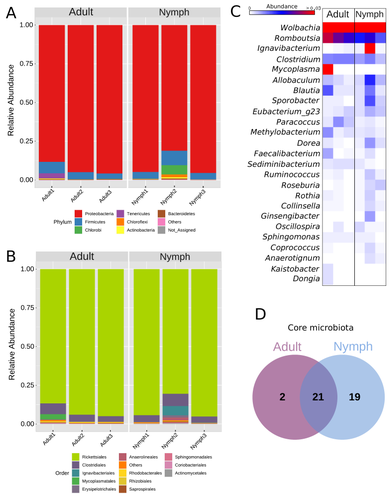
Diversity of bacterial symbionts associated with the tropical plant bug Monalonion velezangeli (Hemiptera: Miridae) revealed by high-throughput 16S-rRNA sequencingShedding light on bacteria associated with an agricultural pest, the tropical plant bug Monalonion velezangeli: a foundational descriptive studyRecommended by Jean-Marie Volland based on reviews by 2 anonymous reviewers
The paper "Diversity of bacterial symbionts associated with the tropical plant bug Monalonion velezangeli (Hemiptera: Miridae) revealed by high-throughput 16S rRNA sequencing" by Navarro-Escalante et al. (2023) is a valuable contribution to entomological research, particularly in the context of pest management. This descriptive study, while not delving into the functional characterization of the associated bacterial strains, lays an essential groundwork for understanding the bacterial components of the microbiota of this agricultural pest. This study is interesting because it provides new information on insect microbiota, especially in a family for which the knowledge of the diversity of bacterial symbionts is very limited. One of the study's core strengths lies in its exploration and definition of the core microbiota of M. velezangeli, which could serve as a foundation for future research aimed at pest control strategies. The use of 16S rRNA sequencing, despite its known limitations, has enabled the profiling of these bacterial communities. The paper highlights the absence of differences in the bacterial communities associated with the nymph and adult stages of the pest, indicating a stable association of these microbes throughout the insect's life cycle. A standout point in the study is the overwhelming presence of the symbiont Wolbachia, accounting for approximately 92% of the bacterial composition. However, intriguingly, the authors also note the absence of Wolbachia in some individuals, suggesting a more complex dynamic that warrants further investigation. This finding is particularly noteworthy, as it opens up questions about the role of Wolbachia and its impact on the biology and ecology of M. velezangeli. The researchers have carefully addressed all the reviewers’ comments and suggestions. They also addressed a potential bias in their study - the overwhelming presence of Wolbachia - by analyzing the bacterial community after the removal of Wolbachia sequences. This careful approach enriches the study's credibility and ensures a more accurate representation of the pest's microbiota. The identification of potentially culturable strains within the core microbiome represents an interesting perspective of this research. This information could be used in future efforts to develop pest control strategies, particularly those employing paratransgenic approaches. The possibility of manipulating these culturable strains to combat M. velezangeli presents an exciting avenue for sustainable pest management. While the study does not investigate the localization of these associated bacteria, whether in the gut or elsewhere, including potentially in dedicated symbiotic organs, it nevertheless offers a valuable descriptive account. This baseline knowledge will be useful for any subsequent functional or localization studies, which could further unravel the complex interactions between M. velezangeli and its microbial partners. In conclusion, the work of Navarro-Escalante et al. is a notable effort to set the stage for future research into the biology of M. velezangeli and its associated microbiota. The findings from this study provide a good reference point for further investigations aimed at pest's biology and exploring innovative pest control strategies. It also represents a valuable contribution to understanding the basic biology of insect-bacteria interactions.
| Diversity of bacterial symbionts associated with the tropical plant bug *Monalonion velezangeli* (Hemiptera: Miridae) revealed by high-throughput 16S-rRNA sequencing | Lucio Navarro-Escalante, Pablo Benavides, Flor Edith Acevedo | <p>Insects and microbes have developed complex symbiotic relationships that evolutionarily and ecologically play beneficial roles for both, the symbiont and the host. In most Hemiptera insects, bacterial symbionts offer mainly nutritional, defensi... | | Microbial ecology and environmental microbiology, Microbial symbiosis | Jean-Marie Volland | 2022-10-31 20:31:54 | ||
29 May 2024
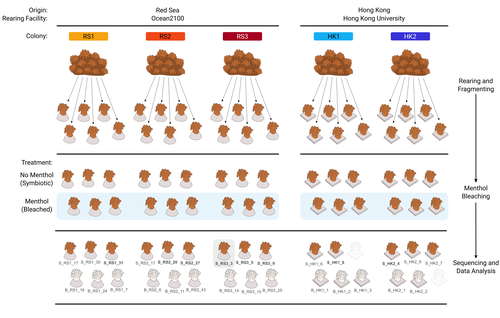
The bacterial microbiome of symbiotic and menthol-bleached polyps of long-term aquarium-reared Galaxea fascicularisAn important step forward in deciphering coral symbiosis through manipulative approachesRecommended by Yui Sato based on reviews by Tony Robinet and 1 anonymous reviewer
As complex multipartite interactions among the coral host and coral-associated microbial entities including the dinoflagellate symbionts, bacteria, archaea and viruses, have been appreciated, a manipulatable, less-complex study system is desired to deepen our functional understanding of this fascinating symbiotic system. Among experimental manipulation approaches, removal of the algal symbionts using menthol is widely implemented; however, its effect on the rest of the coral-associated symbiotic members has not been explored, which is critical knowledge to assess experimental works using this popular method. This preprint by Puntin et al. (https://doi.org/10.1101/2023.08.23.554380) presents an important observation in this aspect. Their initial observations suggest that menthol-induced coral bleaching introduces stochastic changes in associated bacterial communities, which resemble dysbiosis, making bacterial communities more dissimilar from each other. They also observed low taxonomic diversity in bacterial communities on the corals maintained in aquaria over several months, worth noting as a positive value as an experimental system. Their data are preliminary by nature, while they present intriguing ideas that warrant further studies. Reference Puntin G, Wong JCY, Röthig T, Baker DM, Sweet M, Ziegler M (2024). The bacterial microbiome of symbiotic and menthol-bleached polyps of long-term aquarium-reared Galaxea fascicularis (2024). bioRxiv, ver.4., peer-reviewed and recommended by Peer Community In Microbiology. https://doi.org/10.1101/2023.08.23.554380
| The bacterial microbiome of symbiotic and menthol-bleached polyps of long-term aquarium-reared *Galaxea fascicularis* | Giulia Puntin, Jane C.Y. Wong, Till Roethig, David M. Baker, Michael Sweet, Maren Ziegler | <p>Coral reefs support the livelihood of half a billion people but are at high risk of collapse due to the vulnerability of corals to climate change and local anthropogenic stressors. While understanding coral functioning is essential to guide con... | | Microbial symbiosis, Microbiomes | Yui Sato | 2023-08-26 04:50:01 | ||
10 May 2024
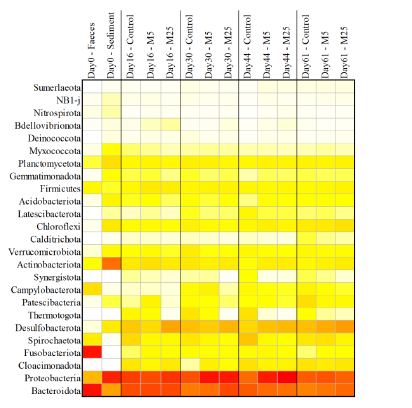
Molybdate delays sulphide formation in the sediment and transfer to the bulk liquid in a model shrimp pondAddition of molybdate to shrimp ponds is a promising new technique to delay the accumulation of toxic H2SRecommended by Roey Angel based on reviews by 2 anonymous reviewers
Shrimp aquaculture ponds are an established technology that helps answer the demand for high-protein food while reducing the impact of fishing on the oceans. However, as a closed system, high in organic matter, aquaculture ponds in general and those used for shrimp in particular tend to develop anoxic sediments and favour sulfate reduction to H2S. The development of hydrogen sulphide, in return, is toxic to the shrimp and can lead to lower yields. A standard solution to the problem is to inject air into the sediments. However, this solution requires additional infrastructure, is costly to operate, and can also disturb other essential life forms in the pond, such as benthic plants. In this work by Torun et al. (2024), the authors used a carefully designed lab model of shrimp ponds to show that the addition of molybdate at concentrations as low as 5 mg/l delayed the accumulation of H2S and pushed the zone rich in sulphide deeper into the sediment. The postulated mechanism for the inhibition in H2S production is that molybdate binds to the ATP sulfurylase in sulphate-reducing bacteria (SRB), and together with ATP, they generate adenosine 5′-phosphosulfate (APS) that cannot be used as an electron acceptor. Surprisingly, however, the growth of SRB was stimulated rather than inhibited in this experiment. While the exact cause remains unknown, the authors postulate that SRB resorted to alternative metabolic pathways such as fermentation. Overall, while this work was done on a model system in the lab, adding molybdate to shrimp aquaculture ponds is a promising technique and should be tested on a larger scale. Reference Torun F, Hostins B, Schryver PD, Boon N, Vrieze JD. (2024). Molybdate delays sulphide formation in the sediment and transfer to the bulk liquid in a model shrimp pond. bioRxiv, ver.3, peer-reviewed and recommended by Peer Community In Microbiology. https://doi.org/10.1101/2023.11.16.567380 | Molybdate delays sulphide formation in the sediment and transfer to the bulk liquid in a model shrimp pond | Funda Torun, Barbara Hostins, Peter De Schryver, Nico Boon, Jo De Vrieze | <p>Shrimp are commonly cultured in earthen aquaculture ponds where organic-rich uneaten feed and faeces accumulate on and in the sediment to form anaerobic zones. Since the pond water is rich in sulphate, these anaerobic conditions eventually lead... | | Microbial biotechnology, Microbial ecology and environmental microbiology, Microbiomes | Roey Angel | 2023-11-20 12:08:51 | ||
25 Apr 2023
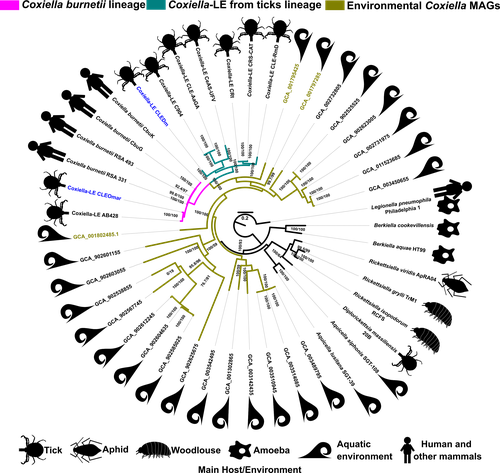
Genomic Changes During the Evolution of the Coxiella Genus Along the Parasitism-Mutualism Continuum.Lifestyle transitions in endosymbiosisRecommended by Daniel Tamarit based on reviews by Sophie Abby, Adam Ossowicki and 1 anonymous reviewerHost-microbe symbioses are an essential component of many ecological systems, playing critical roles in the physiology and evolution of all involved partners. In this context, the bacterial family that includes Coxiella burnetii, the causative agent of Q fever, is of particular interest. The Coxiellaceae family is a complex group with members that have adopted a variety of specializations. Closely related lineages to C. burnetii are tick mutualists (Coxiella-like endosymbionts) and aquatic bacteria that may include both free living and symbiotic species. Additionally, four related genera within this family include symbionts of insects and amoebae. Exactly how and when pathogenicity and mutualism evolved in this lineage is not clear, thus remaining a valuable line of enquiry that can help establish general principles on these lifestyle transitions. A new study by Santos-Garcia and colleagues (2023) places the spotlight on this bacterial group, obtaining new insights through comparative genomics. The authors add two genomes, one of them a circular contig representing a highly reduced (0.9 Mb) chromosome, that increase the resolution of key branches in the Coxiella evolutionary tree. These include a sister group to C. burnetii and the group immediately subtending them, both entirely containing Coxiella-like endosymbionts. By analyzing genetic potential for metabolism, cell dimorphism, virulence and acidophily, the authors find evidence for the ancestrality of genes associated with a pathogenic lifestyle, and support a scenario by which mutualism arose multiple times in a parasitic lineage. In this context shines a pathogenicity island acquired in the common ancestor of this group and subsequently eroded in mutualistic lineages. This scenario highlights the importance of pre-adaptations that facilitate evolutionary specializations, such as the capabilities for B vitamin biosynthesis (key feature in the adaptation to a mutualistic relationship with organisms with B-vitamin-poor diets) and pH homeostasis (harnessed by C. burnetii for infection). Microbial groups at the crossroads of parasitism and mutualism help us understand the mechanisms underpinning these evolutionary strategies (see e.g. Drew et al, 2021). Transitions in endosymbiosis, including shifts in the parasitism-mutualism continuum, adaptation to new partners, or switches between free-living and host-associated lifestyles, affect the structure of ecological networks, and understanding them can yield crucial insights into how to manipulate microbial symbioses for health outcomes, sustainable agriculture or ecosystem conservation. The Coxiellaceae, by including a diverse set of mutualistic, parasitic and possibly free-living lineages, are a fantastic model group to tackle these questions. Together with other host-associated bacteria, such as Sodalis (Clayton et al, 2012) or Pantoea (Walterson and Stavrinides, 2015) species, these ecologically diverse microbes are valuable assets in the quest to decipher the molecular basis of lifestyle transitions in endosymbiosis. REFERENCES Clayton, A.L., et al (2012). A novel human-infection-derived bacterium provides insights into the evolutionary origins of mutualistic insect–bacterial symbioses. PLoS Genetics, 8: e1002990. https://doi.org/10.1371/journal.pgen.1002990 Drew, G.C., Stevens, E.J., King, K.C. (2021). Microbial evolution and transitions along the parasite-mutualist continuum. Nature Reviews Microbiology, 19: 623-638. https://doi.org/10.1038/s41579-021-00550-7 Santos-Garcia, D., et al. (2023) Genomic changes during the evolution of the Coxiella genus along the parasitism-mutualism continuum. bioRxiv, 2022.10.26.513839, ver. 4 peer-reviewed and recommended by Peer Community In Microbiology. https://doi.org/10.1101/2022.10.26.513839 Walterson, A.M., Stavrinides, J. (2015). Pantoea: insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiology Reviews, 39: 968-984. https://doi.org/10.1093/femsre/fuv027 | Genomic Changes During the Evolution of the Coxiella Genus Along the Parasitism-Mutualism Continuum. | Diego Santos-Garcia, Olivier Morel, Hélène Henri, Adil El Filali, Marie Buysse, Valérie Noël, Karen D. McCoy, Yuval Gottlieb, Lisa Klasson, Lionel Zenner, Olivier Duron, Fabrice Vavre | <p style="text-align: justify;">The Coxiellaceae family is composed of five genera showing lifestyles ranging from free-living to symbiosis. Among them, <em>Coxiella burnetii </em>is a well-known pathogen causing Q fever in humans. This bacterium ... | | Bioinformatics dedicated to microbial studies, Genomic and evolutionary studies, Microbe-microbe and microbe-host interactions, Microbial symbiosis | Daniel Tamarit | 2022-10-27 12:55:14 | ||
17 Aug 2023

Within-species variation in the gut microbiome of medaka (Oryzias latipes) is driven by the interaction of light intensity and genetic backgroundGetting closer to the host-microbe evolutionary relationshipRecommended by Konstantinos (Kostas) Kormas based on reviews by Laetitia Wilkins, Marco Basili and 1 anonymous reviewer
The issue of whether there is a clear and detectable relationship -either deterministic or stochastic- of fish gut microbiota with evolutionary processes is far from being resolved. Studies on fish microbiota are more perplexed as this animal group includes species both from wild and farmed populations (for food production, ornamental fish and animal models), with variable life cycles and ecophysiologies, and all these features expand the type of interactions to be studied. Based on this biological features variability, multiple methodological limitations, especially for the species with wild populations, are perhaps among of the central reasons for this knowledge gap. Therefore, experimental approaches, which can eliminate some of this variability, seem to be the best approach. The preprint by Evangelista et al. (2023) entitled "Within-species variation in the gut microbiome of medaka (Oryzias latipes) is driven by the interaction of light intensity and genetic background" is an example of such a targeted study with a freshwater fish species. Due to the paper's finely detailed experimental design, the interdisciplinary skills of the participating co-authors and exhaustive data analysis, this paper manages to draw solid and reproducible results and conclusions. This renders it not only an insightful contribution towards the more general host-microbe interactions in an evolutionary framework, but also a perfect example on how current and future relevant research should be conducted. I feel confident that this paper will assist other scientits of the field to move forward with their current working hypotheses but also to generate novel ones. Reference : Evangelista C, Kamenova S, Diaz Pauli B, Sandkjenn J, Vollestad A, Edeline E, Trosvik P, de Muinck E (2023) Within-species variation in the gut microbiome of medaka (Oryzias latipes) is driven by the interaction of light intensity and genetic background. bioRxiv, 2023.02.17.528956, ver. 2 peer-reviewed and recommended by Peer Community in Microbiology. https://doi.org/10.1101/2023.02.17.528956 | Within-species variation in the gut microbiome of medaka (*Oryzias latipes*) is driven by the interaction of light intensity and genetic background | Charlotte Evangelista, Stefaniya Kamenova, Beatriz Diaz Pauli, Joakim Sandkjenn, Leif Asbjørn Vøllestad, Eric Edeline, Pål Trosvik, Eric Jacques de Muinck | <p style="text-align: justify;">Unravelling evolution-by-environment interactions on the gut microbiome is particularly relevant considering the unprecedented level of human-driven disruption of the ecological and evolutionary trajectories of spec... | | Microbiomes | Konstantinos (Kostas) Kormas | 2023-03-30 16:53:31 |




